| Up Contents | ||||||||||||||||||
How to fold protein?(More in Abalone project)Although the problem of protein folding is far from being solved in general terms, this process can be simulated for simple stable proteins. It is exemplified by small alpha (tryptophan cage) protein. Trp-Cage was shown to fold in several µs in experiment. Explicit water simulations give similar results. Unfortunately, such simulations require huge calculation time. The calculation time can be reduced by several orders of magnitude by using implicit water models. This interferes with the calculation accuracy, but simulation can be performed on a personal computer. Abalone supports several force fields applicable to numerical experiments on protein folding. The table below shows the force fields that allowed us to fold the above-mentioned protein into native conformation.
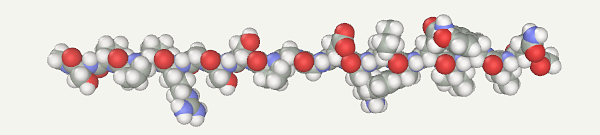
Trp-CageFirst, a protein model should be generated. Open the Chain Builder (Build > Chain menu). Select Amino Acids. Input the Trp-Cage sequence (NLYIQWLKDGGPSSGRPPPS) and press the Apply button.
Select the AMBER94 force field (Build > Assing FF types menu AMBER94) and make sure that the implicit water model is enabled (Build > Implicit water menu). Atoms of amino acid side radicals can overlap during the assembly. Such overlapping can be obviated by a preliminary 20-step optimization. Start optimization by clicking Compute > Remove clashes menu. Apart form new models, short optimization is recommended after the force field is changed, since the model can appear in a high-energy state in terms of another force field. Accordingly, the force field was specified before the optimization.
After the initial model was generated, a molecular dynamics simulation can be
started. Invoke the Compute > Dynamics panel. Set the temperature
to 350 K and simulation time to 10 000 ps (10 ns),
and start the process by Run button
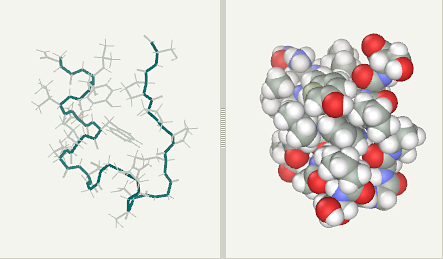
Trp-Cage experimental (pdb1L2Y NMR)
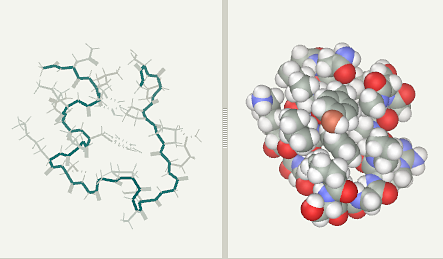
Trp-Cage folding simulation after 2 ns (file)
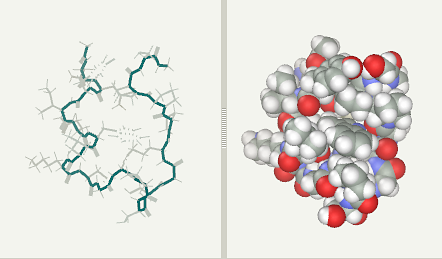
Trp-Cage folding simulation after 4 ns (file)
|
||||||||||||||||||
| Up Contents | ||||||||||||||||||
| Copyright © 2006-2009 Agile Molecule. All rights reserved | ||||||||||||||||||